自身免病(autoimmune diseases)是指机体对自身抗原发生免疫反应而导致自身组织损害所引起的疾病。自从Donath 与Landsteiner提出此概念以来,许多疾病相继被列为自身免疫性疾病,值得提出的是,自身抗体的存在与自身免疫性疾病并非两个等同的概念,自身抗体可存在于无自身免疫性疾病的正常人特别是老年人,如抗甲状腺球蛋白、甲状腺上皮细胞、胃壁细胞、细胞核DNA抗体等。有时,受损或抗原性发生变化的组织可激发自身抗体的产生,如心肌缺血时,坏死的心肌可导致抗心肌自身抗体形成,但此抗本并无致病作用,是一种继发性免疫反应。因此,要确定自身免疫性疾病的存在一般需要根据:①有自身免疫反应的存在,②排除继发性免疫反应之可能,③排除其他病因的存在。
一、自身免疫性疾病的发病机制
自身免疫病的确切发病机制不明,可能与下列因素有关:
1.免疫耐受的丢失 对特异性抗原不产生免疫应答的状态称免疫耐受。通常机体对自身抗原是耐受的,下列情况可导致失耐受:
(1)抗原性质变异:机体对于原本耐受的自身抗原,由于物理、化学药物、微生物等因素的是影响而发生变性、降解,暴露了新的抗原决定簇。例如变性的γ-球蛋白因暴露新的抗原决定簇而获得抗原性,从而诱发自身抗体(类风湿因子)。或通过修饰原本耐受抗原的截体部分,从而回避了TH细胞的耐受,导致免疫应答。这是由于大部分的自身抗原属于一种半抗原和载体的复合体,其中B细胞识别的是半抗原的决定簇,T细胞识别的是载体的决定簇,引起免疫应答时二种信号缺一不可,而一般机体对自身抗原的耐受性往往只是限于T细胞,如载体的抗原决定簇经过修饰,即可为T细胞识别,而具有对该抗原发生反应潜能的B细胞一旦获得TH的信号,就会分化、增殖,产生大量自身抗体。
(2)交叉免疫反应:与机体某些组织抗原成分相同的外来抗原称为共同抗原。由共同抗原刺激机体产生的共同抗体,可与有关组织发生交叉免疫反应,引起免疫损伤。例如A组B型溶血性链球菌细胞壁的M蛋白与人体心肌纤维的肌膜有共同抗原,链球菌感染后,抗链球菌抗体可与心肌纤维发生交叉反应,引起损害,导致风湿性心肌炎。
2.免疫反应调节异常 TH细胞和T抑制细胞(TS)对自身反应性B细胞的调控作用十分重要,当Ts细胞功能过低或TH细胞功能过度时,则可有多量自身抗体形成。已知在NZB/WF1小鼠中随着鼠龄的增长Ts细胞明显减少,由于Ts细胞功能的过早降低,出现过量自身抗体,诱发与人类系统性红斑狼疮(SLE)类似的自身免疫性疾病。
3.遗传因素 自身免疫病与遗传因素有较密切的关系,下列事实可说明这一情况:①很多自身免疫病如SLE、自身免疫性溶血性贫血、自身免疫性甲状腺炎等均具有家族史。②有些自身免疫病与HLA抗原表达的类型有联系,例如人类强直性脊柱炎与HLA-B27关系密切,已有报道将HLA-B27基因转至大鼠,转基因大鼠即可诱发强直性脊柱炎。
4.病毒因素 病毒与自身免疫病的关系已在小鼠的自发性自身免疫病中得到一些证明,例如NZB小鼠的多种组织中有C型病毒及其抗原的存在,在病变肾小球沉积的免疫复合物中也有此类抗原的存在。病毒诱发自身免疫病的机制尚未完全清楚,可能是通过改变自身抗原载本的决定簇而回避了T细胞的耐受作用;也可能作为B细胞的佐剂(如EBV)促进自身抗体形成;或感染、灭活Ts细胞,使自身反应B细胞失去控制,产生大量自身抗体。此外,有些病毒基因可整合到宿主细胞的DNA中,从而引起体细胞变异(不能被识别)而引起自身免疫反应。
自身免疫性疾病往往具有以下共同特点:①患者有明显的家族倾向性,不少与HLA抗原尤其是与D/DR基因位点相关,女性多于男性;②血液中存在高滴度自身抗体和(或)能与自身组织成分起反应的致敏淋巴细胞;③疾病常呈现反复发作和慢性迁延的过程;④病因大多不明,少数由药物(免疫性溶血性贫血、血小板减少性紫癜)、外伤(交感性眼炎)等所致;⑤可在实验动物中复制出类似人类自身免疫病的模型。
二、自身免疫性疾病的类型和举例
自身免疫性疾病可分为二大类:
(一)器官特异性自身免疫病
组织器官的病理损害和功能障碍仅限于抗体或致敏淋巴细胞所针对的某一器官。主要有慢性淋巴性甲状腺炎、甲状腺功能亢进、胰岛素依赖型糖尿病、重症肌无力、慢性溃疡性结肠炎、恶性贫血伴慢性萎缩性胃炎、肺出血肾炎综合征(goodpasture syndrome)、寻常天疱疮、类天疱疮、原发性胆汁性肝硬变、多发性脑脊髓硬化症、急性特发性多神经炎等,其中常见者将分别于各系统疾病中叙述。
(二)系统性自身免疫病
由于抗原抗体复合物广泛沉积于血管壁等原因导致全身多器官损害,称系统性自身疫病。习惯上又称之为胶原病或结缔组织病,这是由于免疫损伤导致血管壁及间质的纤维素样坏死性炎及随后产生多器官的胶原纤维增生所致。事实上无论从超微结构及生化代谢看,胶原纤维大多并无原发性改变,以下简述几种常见的系统性自身免疫病。
1.系统性红斑狼疮(systemic lupus erythematosus, SLE) 是一种比较常见的系统性自身免疫病,具有以抗核抗体为主的多种自身抗体和广泛的小动脉病变及多系统的受累。临床表现主要有发热,皮损(如面部蝶形红斑)及关节、肾、肝、心浆膜等损害,以及全血细胞的减少。多见于年轻妇女,男女比为1:6~9,病程迁延反复,预后差。
病因与发病机制 本病的病因和发病机制不明,目前的研究主要集中在以下三个方面。
(1)免疫因素:患者体内有多种自身抗体形成,提示B细胞活动亢进是本病的发病基础。周围血中B细胞体外培养实验结果发现其增殖能力较正常强8~10倍。
(2)遗传因素:遗传因素与本病的关系表现为:①在纯合子双胎中有很高(69%)的一致性,②SLE患者家属成员中发病的可能性明显增加,③北美白人中SLe 与HLA DR2、DR3有关。这可能是由于位于HLA D区的免疫反应基因(Ir)对抗原(包括自身抗原)所激发的免疫反应的程度有调节作用的缘故。
(3)其他:非遗传因素在启动自身免疫反应中亦起着一定的作用。这些因素包括:①药物:盐酸肼苯哒嗪(hydralazine)、普鲁卡因酰胺(procainamide)等可引起SLE样反应。但停药后常可自愈;②病毒:在实验动物NZB和NZB/WF1小鼠中的自发性SLE样病中发现C型病毒感染,在肾小球中可检出病毒抗原-抗体复合物。但在SLE病中病毒因素尚未能充分得到证实;③性激素对SLE的发生有重要影响,其中雄激素似有保护作用,而雌激素则似有助长作用,故患者以女性为多,特别多发生在生育年龄,病情在月经和妊娠期加重。
自身抗体及组织损害机制本病患者体内有多种自身抗体,95%以上病人抗核抗体阳性,可出现抗DNA(双股、单股)、抗组蛋白、抗RNA-非组蛋白、抗核糖核蛋白(主要为Smith抗原)、抗粒细胞、抗血小板、抗平滑肌等抗体,其中抗双股DNA和抗Smith抗原具相对特异性,阳性率分别为60%和30%,而在其他结缔组织病的阳性率,均低于5%。
抗核抗体并无细胞毒性,但能攻击变性或胞膜受损的粒细胞,一旦它与细胞核接触,即可使胞核肿胀,呈均质状一片,并被挤出胞体,形成狼疮(LE)小体,LE小体对中性粒细胞、巨噬细胞有趋化性(图4-8),在补体存在时可促进细胞的吞噬作用。吞噬了LE小体的细胞为狼疮细胞(图4-9)。在组织中,LE小体呈圆或椭圆形,HE染色时苏木素着色而蓝染,故又称苏木素小体,主要见於肾小球或肾间质。一般仅在20%的患者可检见苏木素小体,为诊断SLE的特征性依据。
SLE的组织损害与自身抗体的存在有关,多数内脏病变是免疫复合物所介导(Ⅲ型变态反应),其中主要为DNA-抗DNA复合物所致的血管和肾小球病变,其次为特异性抗红细胞、粒细胞、血小板自身抗体经Ⅱ型变态反应导致相应血细胞的损害和溶解,引起全贫血。
病变急性坏死性小动脉、细动脉炎是本病的基础病变,几乎存在于所有患者并累及全身各器官。活动期病变以纤维素样坏死为主。慢性期血管壁纤维化明显,管腔狭窄,血管周围有淋巴细胞浸润伴水肿及基质增加。有时血管外膜纤维母细胞增生明显,胶原纤维增多,形成洋葱皮样结构,以脾中央动脉的变化最为突出。应用免疫组织化学方法可证实受累的血管壁中有免疫球蛋白、补体、纤维蛋白、DNA等存在,提示有抗原-抗体复合物机制的参与。
(1)肾:肾功能衰竭是SLE的主要死亡原因。SLE病人几乎均有不同程度的肾损害,约60%病便以狼疮性肾炎为主要表现.常见的类型有系膜增生型(10%~15%)、局灶增生型(10%~15%)、弥漫增生型(40%~50%)和膜型(10%~20%)。各型狼疮性肾炎的病变,类同于相应的原发性肾小球肾炎,各型病变间常有交叉,因此肾小球的病变呈多样性,晚期可出现典型的硬化性肾炎的表现。肾炎病变的发生主要基于肾小球中免疫复合物的沉积,可位于系膜区、内皮下和上皮下。其中弥漫增生型狼疮性肾炎中内皮下大量免疫复合物的沉积,是SLE急性期的特征性病变。在弥漫增生型及膜型病例中,约半数病例在间质及肾小管基膜上亦有免疫复合物沉积,因此肾小球病变和间质的炎症反应在狼疮性肾炎中十分明显(图4-10)。苏木素小体的出现有明确的诊断意义。

图4-8 红斑性狼疮花形细胞簇
游离的LE小体被多个粒细胞包围,形成花形细胞族(采自 Dubois)
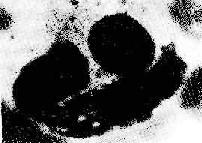
图4-9 狼疮细胞
胞浆内吞噬了两个LE小体,胞核被挤在一边
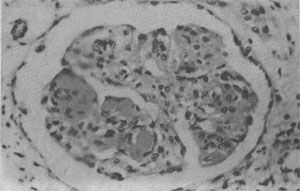
图4-10 狼疮性肾小球肾炎
肾小球毛细血管丛节段性纤维素样坏死,伴系膜细胞增生;间质炎细胞浸润
(2)皮肤:约40%的SLE病人有明显皮肤损害,以面部蝶形红斑最为典型,亦可累及躯干和四肢。镜下,表皮常有萎缩、角化过度、毛囊角质栓形成、基底细胞液化,表皮和真皮交界处水肿,基度膜、小动脉壁和真皮的胶原纤维可发生纤维素样坏死,血管周围常有淋巴细胞浸润。免疫荧光证实真皮与表皮交界处有IgG、IgM及C3的沉积,形成颗粒或团块状的荧光带即“狼疮带”,可能是坏死上皮细胞释出之抗原与血循环中弥散出来的抗核抗体等自身抗体形成的免疫复合物。狼疮带的出现对本病有诊断意义。
(3)心:大约半数病例有心脏受累,心瓣膜非细菌性疣赘性心内膜炎(nonbacterial verrucous endocarditis或Libman-Sach endocarditis)最为典型,敖生物常累及二尖瓣或三尖瓣,其特点为:大小自1mm至3~4mm,数目单个或多个不等,分布极不规则,可累及瓣膜之前后面或心腔之内膜或腱索(图4-11)。镜下,赘生物由纤维蛋白和坏死碎屑及炎症细胞构成,根部基质发生纤维素样坏死,伴炎细胞浸润,后期发生机化。
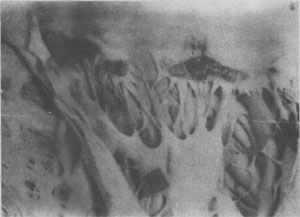
图4-11 红斑性狼疮的心瓣膜疣状心内膜炎
二尖瓣上有疣状赘生物形成
(4)关节:90%以上的病例有不同程度的关节受累。滑膜充血水肿,有较多单个核细胞浸润。于紧接上皮浅表部位的结缔组织可出现灶性纤维素样坏死,但很少侵犯关节软骨等深部组织,因此极少引起关节畸形。
(5)肝:约25%的病例可出现肝损害,称狼疮性肝炎。可表现为汇管区及汇管区周围的单个核细胞浸润及附近肝细胞的碎屑状坏死等慢性活动性肝炎的典型病变。亦可仅有少量散在分布的小灶性坏死等轻微病变。
(6)脾:体积略增大,包膜增厚,滤泡增生颇常见。红髓中有多量浆细胞,内含IgG、IgM,最突出的变化是小动脉周围纤维化,形成洋葱皮样结构(图4-12)。
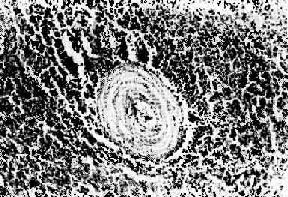
图4-12 红斑性狼疮之脾病变
脾小体中央细动脉壁呈洋葱皮样结构
(7)淋巴结:全身淋巴结均有不同程度的肿大,窦内皮增生。其中较多的浆细胞,小血管变化与脾所见相同。
2.口眼干燥综合征 口眼干燥综合症(Sjǒgren syndrome)临床上表现为眼干、口干等特征,乃唾液腺、泪腺受免疫损伤所致。本病可单独存在,也可与其他自身免疫病同时存在,后者最常见的是类风湿性关节炎、SLE等。病变主要累及唾液腺及泪腺,其他外分泌腺包括呼吸道、消化道腺体也可受累。
唾液腺的组织学病变主要表现为腺管周围大量炎细胞浸润,主要是淋巴细胞和浆细胞,有时可形成淋巴滤泡并有生发中心形成。伴腺管上皮增生,引起管腔阻塞。病变晚期腺泡萎缩、纤维化,为脂肪组织所替代。个别病例浸润的淋巴细胞形成淋巴瘤样结构。由于唾液腺的破坏而引起口腔粘膜干裂及溃疡形成。
泪腺的类似病变可导致角膜上皮干燥、炎症及溃疡形成。呼吸道、消化道受累可导致相应的鼻炎、喉炎、支气管炎、肺炎及萎缩性胃炎。在肾可发生间质性肾炎,肾小管周围大量单个核细胞浸润,导致肾小管萎缩、纤维化,因肾小管功能损害而引起肾小管性酸中毒、磷酸盐尿等颇常见。
淋巴结肿大并有增生性变化,核分裂多,故又名假性淋巴瘤。值得提出的是本病患者发生恶生淋巴瘤的机会较正常人高40倍。
发病机制 本病的发病机制尚不清楚。由于常伴发SLE和类风湿性关节炎,提示本病的发生与免疫性损伤有关。患者B细胞功能过度,表现为多克隆高球蛋白血症和类风湿因子(RF)、抗核抗体、冷球蛋白及抗唾液腺抗体的形成。近年来并发现两种特征性抗核糖核蛋白成分的自身抗体,分别命名为抗SS-B和SS-A,在本病有很高的阳性率(60%、70%),对本病的诊断有参考价值。病灶处有大量B及T细胞浸润,后者大部分为T辅助细胞,也有一部分为T杀伤细胞,提示亦有细胞免疫机制的参与。
3.类风湿性关节炎 详见骨关节疾病章。
4.硬皮病 硬皮病(scleroderma)又名进行性系统性硬化症(progressive systemic sclerosis),以全身许多器官间质过度纤维化为其特征。95%以上的患者均有皮肤受累的表现;但横纹肌及许多器官(消化道、肺、肾、心等)受累是本病主要损害所在,病变严重者可导致器官功能衰竭,威胁生命。
病因和发病机制本病病因不明,其发病可能与以下因素有关:
(1)胶原合成增加:体外培养证实,患者纤维母细胞合成胶原的能力明显高于正常人,合成超过降解,导致大量胶原纤维的积集;
(2)Ⅳ型变态反应:在皮肤病变中有T细胞浸润,所分泌的淋巴因子及其刺激巨噬细胞分泌的因子可刺激纤维母细胞大量合成胶原;
(3)自身抗体:50%患者有轻度高丙种球蛋白血症及多种自身抗体,包括RF,抗平滑肌抗体,抗核抗体等,可能由于抗原抗体免疫复合物的沉积或内皮细胞毒的作用,造成小血管内皮细胞损伤、血栓形成、管壁纤维化、管腔狭窄,导致组织缺氧而引起纤维间质增生。
【病变】
(1)皮肤:病变由指端开始,向心性发展,累及前臂、肩、颈、脸,使关节活动受限。早期受累的皮肤发生水肿,质韧。镜下,主要表现为小血管周围淋巴细胞浸润,毛细血管内皮细胞肿胀、基膜增厚、管腔部分阻塞,间质水肿,胶原纤维肿胀,嗜酸性增强。随着病变的发展,真皮中胶原纤维明显增加,并与皮下组织紧密结合,表皮萎缩变平,黑色素增加,钉突和附属器萎缩消失,小血管增厚、玻璃样变。晚期手指细而呈爪状、关节活动受限,有时指端坏死甚或脱落,面部无表情呈假面具状。
(2)消化道:约有1/2患者消化道受累,粘膜上皮萎缩,固有层、粘膜下层、肌层为大量胶原纤维所取代,血管周单个核细胞浸润。病变以食管下2/3段最严重,管腔狭窄,缺乏弹性。小肠、结肠也可受累。临床上出现吞咽困难、消化不良等症状。
(3)肾:叶间小动脉病变最为突出,表现为内膜粘液样变性,伴内皮细胞增生及随后的管壁纤维化、管腔明显狭窄,部分病例并有细动脉纤维素样坏死。临床上可出现高血压,与恶性高血压肾病变难以区别。约50%患者死于肾功能衰竭。
(4)肺:弥漫性间质纤维化,肺泡扩张、胞泡隔断裂,形成囊样空腔,本病是造成蜂窝肺的重要原因之一。
5.结节性多动脉炎 结节性多动脉炎(polyarteritis nodosa)是全身动脉系统的疾病,表现为中小动脉壁的坏死性炎症。患者以青年人为多,有时也可发生在儿童及老人、男女之比为2~3:1。
病变各系统或器官的中小动脉均可受累,其中以肾(85%)、心(75%)、肝(65%)、消化道(50%)最为常见。此外,胰、睾丸、骨骼肌、神经系统和皮肤也可受累。
病变多呈节段性,以血管分叉处最为常见。内眼观,病灶处形成直径约2~4mm的灰白色小结节,结节之间的血管壁外观正常。镜下,急性期表现为急性坏死炎症,病变从内膜和中膜内层开始,扩展至管壁全层及外膜周围,纤维素样坏死颇为显著,伴炎细胞浸润(图4-13)尤以嗜酸性及中性粒细胞为多,继而有血栓形成。以后的进展是纤维增生,管壁呈结节性增厚,管腔机化阻塞和明显的动脉周围纤维化。值得注意的是早期炎性坏死变化及后期胶原化可同时存在。病变的主要后果是缺血性损害和梗死形成。
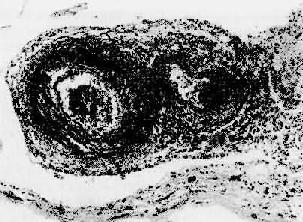
图4-13 结节性多动脉炎
两个动脉壁的各层都有炎性细胞浸润,外膜尤为显著。中膜发生纤维素样坏死
本病病变分布广泛,临床表现变异多端,患者常有低热、乏力、粒细胞增多以及多系统受累的症状,如血尿、肾功能衰竭、高血压、腹痛、腹泻、黑粪及周围神经炎等。病程快慢不一,经免疫抑制治疗,55%患者可存活。
病因与发病机制 病因和发病机制不明,动物实验提示,体液因素在本病的发生中起着重要作用。免疫荧光技术证实,人结节性多动脉炎血管壁中有免疫球蛋白和补体,有些还有HBsAg,约50%患者血清HBsAg或抗HBs阳性。
6.Wegener肉芽肿病 Wegener肉芽肿病是一种少见病,具有以下特点:①小血管急性坏死性脉管炎,可累有各器官的血管,以呼吸道、肾、脾最常受累。表现为小动脉、小静脉管壁的纤维素样坏死,伴弥漫性中性和嗜酸性粒细胞浸润;②呼吸道肉芽肿性坏死性病变,可累及口、鼻腔、鼻旁窦、喉、气管、支气管和肺。病变为由大量积集的单核巨噬细胞、淋巴细胞以及少量多核巨细胞、类上皮细胞、纤维母细胞组成的肉芽肿,中央可陷于成片凝固性坏死。肉眼常形成明显的肿块,表面则因坏死溃破而有溃疡形成;③坏死性肾小球肾炎,表现为在局灶性或弥漫增生性肾小球肾炎的基础上,有节段性毛细血管袢的纤维素样坏死,血栓形成,如未经治疗可发展为快速进行性肾炎,病程凶险,出现进行性肾功能衰竭。
本病的病因不明,由于有明显的血管炎,并于局部可检得免疫球蛋白和补体,提示其发病与Ⅲ型变态反应有关。但呼吸道出现的肉芽肿和坏死性病变,又提示可能与Ⅳ型变态反应有关,临床上应用细胞毒药物大多能使本病缓解。