变性疾病是一组原因不明的中枢神经系统疾病,病变特点在于选择性地累及某1~2个功能系统的神经细胞而引起受累部位特定的临床表现,如累及大脑皮层神经细胞的病变,主要表现为痴呆;累及基底核椎体外运动神经系统则引起运动障碍,累及小脑可导致共济失调等。本组疾病的共同病理特点为受累部位神经元的萎缩、死亡和星形胶质细胞增生,此外不同的疾病还可有各自特殊的病变,如在细胞内形成包含体或发生神经原纤维缠结等病变。几种主要的变性疾病见表16-3。
表16-3 几种主要的变性疾病
| 病变主要累及部位 | 疾 病 |
| 大脑皮质 | Alzheimer病 |
| Pick病 | |
| 基底核及脑干 | Huntington病 |
| 震颤性麻痹 | |
| 纹状体黑质变性 | |
| 进行性核上麻痹 | |
| Shy-Drager综合征 | |
| 脊髓与小脑 | 橄榄桥脑小脑萎缩 |
| Friedreich共济失调 | |
| 共济失调性毛细血管扩张症 | |
| 运动神经元 | 肌萎缩性侧索硬化 |
| 脊髓性肌萎缩 |
一、Alzheimer病
Alzheimer病又称初老期痴呆,是以进行性痴呆为主要临床表现的大脑变性性疾病,起病多在50岁以后。随着人类寿命的延长,本病的发病率呈增高趋势。按照美国的诊断标准,上海60岁以上人群发病率为3.46%。65岁以上人群为4.61%。临床表现为进行性精神状态衰变,包括记忆、智力、定向、判断能力、情感障碍和行为失常甚至发生意识模糊等。患者通常在发病后5~6年内死于继发感染和全身衰竭。
【病理变化】
肉眼观,脑萎缩明显,脑回窄、脑沟宽,病变以额叶、顶叶及颞叶最显着(图16-32),脑切面可见代偿性脑室扩张。
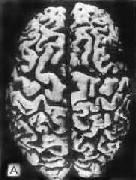
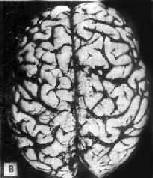
图16-32 初老期痴呆的脑
示脑明显萎缩(A),与正常脑(B)的对比
镜下,本病最主要的组织病变有:老年斑,神经原纤维缠结,颗粒空泡变性,Hirano小体等。
(1)老年斑:为细胞外结构,直径为20~150μm,最多见于内嗅区皮质、海马CA-1区,其次为额叶和顶叶皮质。银染色显示,斑块中心为一均匀的嗜银团,刚果红染色呈阳性反应,提示其中含淀粉样蛋白,其中含该蛋白的前体β/A-4蛋白及免疫球蛋白成分。中心周围有空晕环绕,外围有不规则嗜银颗粒或丝状物质。电镜下可见该斑块主要由多个异常扩张变性之轴索突触终末构成(图16-33)。
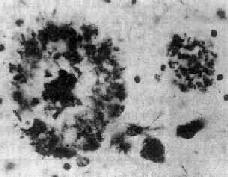
图16-33 老年斑
左侧为典型的老年斑。中心为嗜银团,围以空晕,外围为嗜银性细颗粒及细丝,周围见星形胶质细胞。右侧见一正在形成的老年斑
(2)神经原纤维缠结:神经原纤维增粗扭曲形成缠结,在HE染色中往往较模糊,呈淡蓝色,而银染色最为清楚。电镜下证实其为双螺旋缠绕的细丝构成,多见于较大的神经元,尤以海马、杏仁核、颞叶内侧,额叶皮质的锥体细胞最为多见。此外,前脑底Meynert基底核及蓝斑中也可见到。这一变化是神经元趋向死亡的标志(图16-34)。
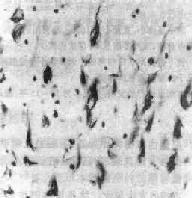
图16-34 神经原纤维缠结
脑皮质锥体细胞神经原纤维缠结增粗呈焰状(Bielschowsky银染色)
(3)颗粒空泡变性:表现为神经细胞胞浆中出现小空泡,内含嗜银颗粒,多见于海马Sommer区的锥体细胞。
(4)Hirano小体:为神经细胞树突近端棒形嗜酸性包含体,生化分析证实大多为肌动蛋白,多见于海马锥体细胞。
上述变化均非特异性,可见于无特殊病变之老龄脑,仅当其数目多并具特定的分布部位时才能作为Alzheimer病的诊断依据。
【病因和发病机制】
病因和发病机制不明。对于本病究竟是一独立的疾病,还是一种加速的老化尚有不少争议。本病的发病可能与下列因素有关:①受教育程度:上海的人群调查资料以及随后世界大多数地区的调查资料证实,本病的发病率与受教育程度有关。文盲及初小文化人群中发病率最高,受到高中以上教育人群中发病率较低。病理研究表明,大脑皮质突触的丧失先于神经元的丧失,突触丧失的程度和痴呆的相关性较老年斑、神经原纤维缠结与痴呆的相关性更加明显。人的不断学习可促进突触的改建,防止突触的丢失。②遗传因素:对初老期痴呆病中具有家属遗传史的病人(遗传性Alzheimer病仅为本病一个少见类型)的研究表明,其控制基因在第21对染色体上,具有多个位点,某些位点与先天愚型(Down症)位点甚接近,因此后者的Alzheimer病的发病率较高。③神经细胞的代谢改变:老年斑中的淀粉样蛋白的前体β/A-4蛋白是正常神经元膜上的一个跨膜蛋白,何以在本病中会发生不溶性沉淀的原因尚待探讨。缠结的神经原纤维中神经微丝,τ蛋白等细胞骨架蛋白呈现过度的磷酸化。某些患者病脑中铝的含量可高于正常。④继发性的递质改变:其中最主要的改变是乙酰胆碱的减少。由于Meynert基底核神经元的大量缺失致其投射到新皮质、海马、杏仁核等区域的乙酰胆碱能纤维减少。综上所述,目前已发现了本病的形态、生化、遗传等各方面的异常改变,但病因和发病机制尚有待阐明。
病理诊断
由于Alzheimer病的病理变化均为非特异性,故必须根据老年斑和神经原纤维缠结的数目及部位,结合患者的年龄和临床表现才能作出判断。参考Khachaturian执笔的美国约定诊断标准(表16-4),并除外其它引起痴呆的原因,如血管源性痴呆以及其它变性疾病等,方可作出诊断。
表16-4 美国诊断Alzheimer病的标准
| 患者年龄 | 老年斑数(×200,/mm2) | 神经原纤维缠结(×200,/mm2) |
| <50岁 | >2~5 | >2~5 |
| ≤60 | >8 | >2~5 |
| ≤75 | >10 | >2~5 |
| >75 | >15 | 有或无 |
二、慢性进行性舞蹈病
慢性进行性舞蹈病又称Huntington舞蹈病,是一种常染色体显性遗传病,突变基因位于第4对染色体。患者的子女中半数可得病,男女患病机会均等,多在20~50岁开始发病。临床表现为舞蹈样动作及进行性痴呆。
脑明显缩小,重量小于1000g,最突出的是两侧尾状核和壳核的萎缩,以致侧脑室明显扩张。大脑皮质特别是额、顶叶萎缩显着,白质也减少。
镜下可见尾状核和壳核中选择性小神经细胞丢失,伴星形胶质细胞增生和胶质纤维化,类似的病变可见于丘脑腹侧核和黑质。
本病呈进行性发展,病程多为10~15年,最后死于并发症。
三、震颤性麻痹
震颤性麻痹(paralysis agitans)又称Parkinson病,是一种缓慢进行性疾病,多发生在50~80岁。临床表现为震颤、肌强直、运动减少、姿势及步态不稳、起步及止步困难、假面具样面容等。
本病的发生与纹状体黑质多巴胺系统损害有关,最主要的是原因不明性(特发性)Parkinson病,其他如甲型脑炎后,动脉硬化,及一氧化碳、锰、汞中毒等,均可产生类似震颤性麻痹症状或病理改变。这些情况统称为Parkinson综合征。
【病理变化】
黑质和蓝斑脱色是本病肉眼变化的特点(图16-35)。镜下可见该处的黑色素细胞丧失,残留的神经细胞中有Lewy小体形成,该小体位于胞浆内,呈圆形,中心嗜酸性着色,折光性强,边缘着色浅。电镜下,该小体由细丝构成,中心细丝包捆致密,周围则较松散。
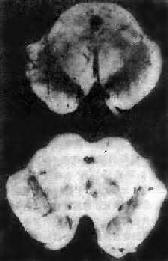
图16-35 Parkinson病
中脑黑质脱色(下),正常中脑黑质完好(上)
由于黑质细胞的变性和脱失,多巴胺合成减少,以致多巴胺(抑制性)与乙酰胆碱(兴奋性)的平衡失调而致病。近年来用左旋多巴(多巴胺的前体)来补充脑组织中多巴胺不足或用抗胆碱能药物以抑制乙酰胆碱的作用,对本病有一定的疗效。
四、肌萎缩性侧索硬化症
肌萎缩性侧索硬化症(amyotrophic lateral sclerosis,ALS)的病变累及锥体束上、下神经元,上神经元在大脑皮质,其轴索经过内囊、脑干及皮质脊髓束与脑运动神经核或脊髓前角下运动神经元相联系。本病发病年龄在40~50岁,男多于女,临床起病缓慢,表现为进行性上、下肢肌萎缩,无力,锥体束损害及脑干运动神经核受损,一般无感觉障碍,病程一般2~6年。
本病的病变多自脊髓向上发展,波及脑干乃至大脑的运动区。下运动神经元变性消失,以颈、腰膨大区最为显着,脊髓前根及其支配的肌肉萎缩。上运动神经元大量变性消失伴星形胶质细胞增生。肉眼观,中央前回萎缩明显。在延髓麻痹型病例中,脑运动神经核特别是第Ⅴ、Ⅸ、Ⅹ、Ⅺ及Ⅻ神经核的神经细胞变性脱失,偶见神经细胞被吞噬,提示持续性神经细胞丢失。残留的神经元皱缩成鬼影状。
运动神经元病的病因和发病机制尚不清楚。野生小鼠自发性运动神经元病系由C型RNA病毒引起,但与人类疾病有无关系尚不清楚。值得注意的是关岛Chamorro族和日本纪伊半岛中本病的发病率高,该两地区土壤中含锰甚高,饮水中钙的含量也高,尸检病例脊髓中含锰量也甚高,因而锰、钙在发病中的作用已受到注意。