脂蛋白脂肪酶(liportein lipase, LPL)是脂肪细胞、心肌细胞、骨骼肌细胞,乳腺细胞以及巨噬细胞等实质细胞合成和分泌的一种糖蛋白,分子量为60kD,含3%~8%碳水化合物。活性LPL以同源二聚体形式存在,通过静电引力与毛细血管内皮细胞表面的多聚糖结合,肝素可以促进此结合形式的LPL释放入血,并可提高其活性。LPL生理功能是催化CM和VLDL核心的TG分解为脂肪酸和单酸甘油酯,以供组织氧化供能和贮存;LPL还参与VLDL和HDL之间的载脂蛋白和磷脂的转换,ApoC-Ⅱ为其必需的辅因子,其中的C端第61~79位氨基酸具有激活LPL的能力。
一、LPL结构与合成
比较不同种类包括人类脂肪组织、牛乳腺、鼠巨噬细胞、猪脂肪组织和禽类的LPL一级结构,发现人类LPL氨基酸序列与哺乳类动物有87%~94%同源性,与禽类比较也有70%同源性,表明LPL在进化过程中的高度保守性。人类LPL、肝脂酶(HL)以及胰脂酶(PL)具有高度相似的氨基酸序列,推测三者可能起源于同一个基因家族,具有共同的作用机制。
目前人类LPL二级结构尚未阐明,推测LPL分子可能由两个结构区域构成:N端区的和C端区,N端区包括1~315位氨基酸,形成一个以β折叠为主的近球形结构,它是LPL重要的功能区,催化活性中心就位于此区。构成LPL催化活性中心的三个氨基酸分别为Ser132、His241和Asp156,用中性氨基酸取代活性中心附近的氨基酸,LPL活性明显下降或消失。Asn43是N端区一重要的糖基化位点,它起着维持LPL正常三维结构的作用。对正常分泌的LPL活性功能具有重要意义。此外,N端区第279~282位和292~309位氨基酸介导LPL与肝素结构。C端区呈一个折叠的柱状连接在球形的N端区,C端区的功能尚有争议,多数认为与介导酶与底物接触,形成活性的LPL同源二聚体以及间接参与酶解过程。
LPL在实质细胞的粗面内质网合成,新合成的LPL留在核周围内质网,属于无活性酶,由mRNA翻译合成的无活性LPL,称为酶前体,再糖基化后,才转化成活性LPL酶,如图5-1所示。从细胞中如何分泌;目前认为有两种机制,其一是细胞合成的LPL后直接分泌,不贮存于细胞内,即称为基本型分泌;其二是调节型分泌,某些细胞新合成LPL贮存于分泌管内,一旦细胞受到一个合适的促分泌剌激。LDL即分泌,此时分泌往往大于合成。所有细胞都具备基本型分泌,只有少部分细胞兼有两种分泌形式。Vannier提出,LPL是结合在插入细胞内分泌器并存在于细胞膜外表面的硫酸肝素糖蛋白(heparin sulphate protoglycans, HSPG),致使酶保持一种无活力的浓缩状态,然后通过一个尚未阐明的机制由肝素促使分泌,即肝素后血浆中得到活化的LPL,分布在含甘油三酯的脂蛋白中,并主要是分解CM和VLDL的甘油三酯并结合附着在这些脂蛋白残粒中,可能形成肝摄取这些颗粒的信号。
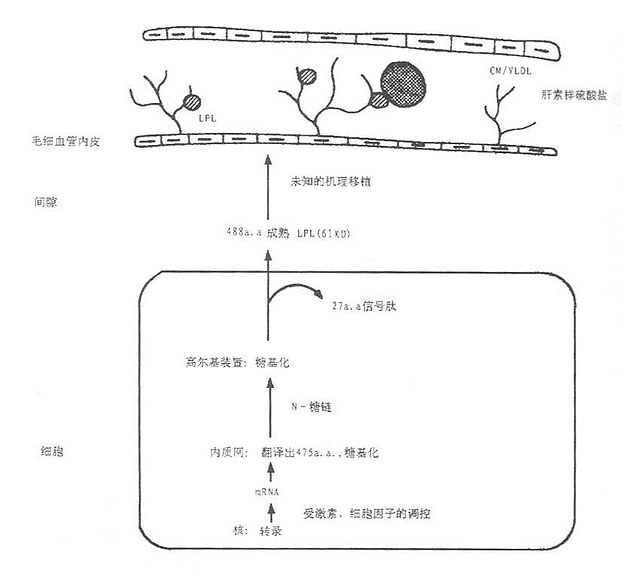
图5-1 LPL的合成
二、LPL基因及其多态性
LPL基因位于8P22,长约35kb,由10个外显子和9个内含子组成,编码475个氨基酸的蛋白质,包括27个氨基酸残基的信号肽。现已查明外显子1长273kp,编码5'端非翻译区和信号肽区。外显子2~5长度分别为161、180、112和234bp,其中外显子2所编码的Asn43糖基化位点为LPL催化活性必需的,外显子4编码脂质结合区。外显子6包含243bp。编码肝素结合区。外显子7~9长度分别为121、183和105bp。编码结构参与与ApoCⅡ结合。外显子10是LPL基因中最大的外显子,长1950bp,编码整个3'端非翻译区。LPL内含子7第序列尚未弄清,报道其中存在Alu重复序列和多态位点。Jurke等研究发现LPL内含子7中有一完整的Alu序列,长282bp,位于内含子7第1027~746位。Delin报道内含子6中也存在Alu序列。Alu序列在人类基因组中约占3%-6%,推测其功能与基因转录的调节,hnRNA的加工以及DNA复制的启动有关,并且是基因重排的热点位置。利用限制性片段长度多态性(RFLPS)技术检测出LPL基因位点存在多态性,主要分布在LPL基因内含子和侧翼序列中,其中内含子6中的PvuⅡ多态位点(图5-2)和内含子8中的HindⅢ(图5-3)多态位点与高脂血症有关,这为高脂血症的家系连锁分析提供遗传标记。LPL基因转录起始点上游730bp区域是转录因子结合部位,5'侧翼区一27位有“TATA”框,是LPL基因启动子所在部位。
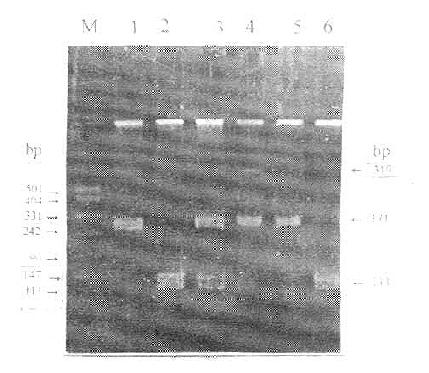
图5-2 LPLPvuⅡ-RFLP
M:DNA marder
1:DNA-PCR产物(319bp)
2-6:分别为P+P+、P+P-、P+P-、P-P-、P--、P+P+、H+H+基因型
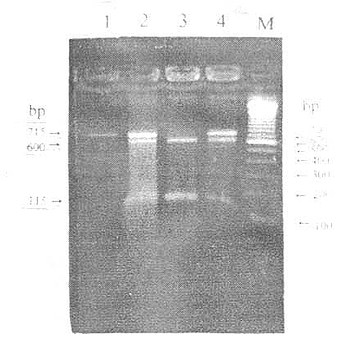
图5-3 LPL HindⅢ-RFLP
M:DNA marker
1~4:分别为H-H-、H-H+、H+H+、H+H-基因型
三、LPL与Ⅰ型高脂血症
Ⅰ型高脂血症为常染色体隐性遗传病,具有家族性。主要临床症状为阵发性腹痛,疹状皮肤黄色瘤和肝脾肿大,生化特征为含高甘油三酯的乳糜微粒在因浆中大量堆积,脂肪耐量显着异常,LPL活性下降,这种患者体内的LPL含量可能完全缺陷,用目前的检测方法测不出LPL存在,也不可能在肝素注射后使血浆LPL活性下降,推测可能是一种异常LPL翻译后修饰所致。
引起Ⅰ型高脂血症常见的LPL基因突变有三类:一是碱基置换突变。按性质又可分为错义突变和无意义突变,错义突变是指基因结构中某个碱基为另一个碱基取代,导致蛋白质分子中相应位置氨基酸改变,这类突变多集中在LPL活性中心所在的N端区,如Gly142、Ala176、Gly188、IIe194、Leu207、Arg243等,这些氨基酸被取代则LPL活性显著降低;无意义突变是指基因编码区发生突变后形成终止密码子,使翻译过程提前终止;导致合成肽链变短,Ⅰ型高脂血症发生与此类突变位点有关,若突变位点在第106位氨基酸的编码基因上,产生出来的短肽链产物不具有LPL催化功能,导致Ⅰ型高脂血症发生;突变位点在第447位氨基酸的编码基因(Ser447-Thr),其产物C端缺失2个氨基酸,不影响LPL活性:二是移码突变。是指在DNA分子中插入或缺失一个或几个核苷酸(但不是3个或3的倍数)造成这一位置以后的一列编码发生移位错误的突变,这种突变有的会生成终止密码而使翻译过程提前终止。有报道1例Ⅰ型高脂血症患者,其G916位碱基发生缺失,产生一个提前终止码,利用Northern印迹技术未检测到可测水平的LPLmRNA,推测此移码突变可能导致LPLmRNA稳定性下降;三是基因重排。表现为大片段缺失或插入,目前发现外显子6中2kb插入,外显子9中3kb缺失,均导致I型高脂血症。
近年研究发现,1例Ⅰ型高脂血症患者脂肪组织中存在正常活性的LPL,肝素后的血浆中检测不出LPL存在,分析其脂肪组织中LPL化学组成,发现这种LPL的寡糖链含有较高水平的果糖,影响了LPL细胞内外的转运,提示翻译后的修饰异常也可影响LPL活性发挥。
脂蛋白脂肪酶基因突变点如表5-1所示。
表5-1 脂蛋白脂肪酶基因突变
| 突变点 | 位 置 | 氨基酸异常 |
| (1)剪接突变 | ||
| GT→AT | 内含子2 | 异常的mRNA生成 |
| AG→AA | 内含子2 | 异常的mRNA生成 |
| (2)错义突变 | ||
| T→C | 外显子3 | Trp96→Arg |
| A→G | 外显子4 | His136→Arg |
| G→A | 外显子4 | Gly142→Clu |
| A→G | 外显子5 | Asp156→Gly |
| G→A | 外显子5 | Asp156→Asn |
| C→G | 外显子5 | Ala157→Arg |
| G→A | 外显子5 | Gly176→Thr |
| G→A | 外显子5 | Gly186→Glu |
| T→C | 外显子5 | Ile194→Thr |
| C→G | 外显子5 | Asp204→Glu |
| C→T | 外显子5 | Pro207→Leu |
| T→A | 外显子5 | Cys216→Ser |
| G→A | 外显子6 | Arg242→His |
| T→A | 外显子6 | Ser244→Thr |
| G→A | 外显子6 | Asp250→Asn |
| T→C | 外显子6 | Tyr252→His |
| (3)无义突变 | ||
| T→A | 外显子3 | Tyr61→Stop |
| C→T | 外显子3 | Gln165→Stop |
| C→(A/G) | 外显子6 | Tyr282→Stop |
| G→A | 外显子6 | Trp282→Stop |
| (4)插入 | ||
| 2kb插入 | 内含子6 | 无效等位基因 |
| 5kb插入 | 外显子3 | 外显子4的stop |
| (5)缺失 | ||
| 6kb的缺失 | 内含子2~5 | 无效等位基因 |
| 1bp的缺失 | 外显子5 | 外显子5的stop |